Compliance Assurance




CrossLab Enterprise Services
A Learning Café
for the Enterprising Lab
Advance operations across your organization
If you have a centralized lab or shared facilities, Agilent CrossLab Enterprise Services can give you insights into improving efficiency, optimizing resources, and increasing instrument uptime. Our asset management, compliance, operational, and digital services work together to simplify, optimize, and transform your lab operations.
Why should your lab make compliance a priority?
Compliance reduces your regulatory risk and allows you to concentrate on your critical science.
The validity of your analytical results depends on many factors, but for laboratories in regulated industries, there is a particular focus on two areas:
- The analytical instrument must be qualified for intended use.
- The software must be validated for intended use.
If these requirements are not satisfied, you may be questioned during audits and inspections. These questions can jeopardize confidence in the integrity of your analytical results. Even worse, they can raise concerns about the data already supplied to regulatory agencies, as well as the quality of your products.
While lab compliance is essential for regulated industries, it is sometimes perceived as a cost to be managed. The real cost, however, relates to the impact of remediation, if deficiencies are identified during regulatory audits and inspections.
Interpreting requirements
Pharmaceutical labs must interpret regulatory requirements such as pharmacopeial general chapters and national good practice (GxP) regulations. Both typically focus on the criteria that must be satisfied and not on the practical details of what needs to be done. This requirement complicates lab compliance, because pharmacopeia and GxP regulations are not harmonized.
The good news
Your lab can simplify audit preparation and reduce regulatory risk. How? By adopting a harmonized approach to instrument qualification and computer system validation that is audit proven and capable of supporting many analytical technologies.
What do changing regulations mean for your lab?
Based on laboratory audit findings, the focus on data integrity continues to evolve. Regulatory agencies worldwide now look more closely at your laboratory operations—particularly, the validity and integrity associated with repeat work, data traceability, and data governance. Laboratories must comply with regulatory requirements against this background of evolving data integrity focus.
This intense focus, however, comes at a time when your lab might be struggling to maximize productivity. Therefore, you must balance the challenges of audit preparation with lab improvement projects and the automation of manual tasks. All are essential, but they compete for the same limited resources.
Audits and investigations
The requirement for regulated laboratories to demonstrate that instruments are suitable for their intended use is still in place. However, the questions that you need to answer during an audit have changed. Regulators are increasingly focusing on:
- Software validation: Have workflows and configurations been validated to remove data integrity risks?
- Data integrity: How was your analytical instrument qualification work performed and documented?
- Compliance with USP <1058>: Does your qualification comply with requirements/frameworks such as USP <1058>?
In a data-integrity audit, the emphasis is on “proving” that your data are not fraudulent rather than solely on scientific justification. That means your qualification must be founded on scientific principles and include both holistic and metrology-based instrument testing. If your lab does not align your AIQ with new <1058> requirements, you will increase your risk of noncompliance.
A globalized regulatory approach
The data integrity focus has led to collaboration, harmonization, and data exchanges between regulatory bodies through organizations like the Pharmaceutical Inspection Co-operation Scheme (PIC/S).
Many regulatory agencies have internal targets and improvement initiatives, such as the FDA data dashboard, that drive global transparency and availability of noncompliance data. The International Council for Harmonization (ICH) is another example of regulatory globalization. Since its 2015 announcement of organizational changes, ICH has grown to include 17 members and 32 observers.
Resources
White paper
Flyer
Recorded seminar
Risk Assessment Guide
How can you comply with USP <1058> AIQ?
Regulated laboratories must demonstrate that analytical instruments are suitable for their intended use. U.S. Pharmacopeia general chapter <1058> (analytical instrument qualification (AIQ)) defines the essential framework that your lab should follow.
As the only major pharmacopeia with a general chapter dedicated to AIQ, <1058> is a key regulatory reference and changes have global implications for instrument qualification.
When surveyed, however, 68% of laboratories were unaware of the differences between the obsolete 2008 version of <1058> and the current (2017) version. Specifically:
- User requirements must be documented.
- Design qualification must document why the instrument is suitable.
- Risk assessment is required.
- Qualification must be tied to user requirements.
- PQ, or performance qualification, testing is now a requirement.
In addition, the 2017 updates to USP <1058> more strongly aligned USP <1058> with GAMP 5 requirements for instrument qualification and software validation.
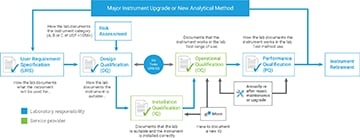
An integrated, life cycle-based approach to AIQ
The Automated Compliance Engine (ACE) software that Agilent uses to provide AIQ services is compliant with USP <1058> requirements. For example, our equipment qualification plans (EQP) can be configured to ensure that operational qualification (OQ) meets user requirements—such as testing the range of use.
Stay compliant, and spend more time on what matters
To help your lab comply with USP <1058> requirements, Agilent has developed four white papers. Through our Compliance Consulting Services, we can also help you implement cost-effective electronic qualification and align your SOPs to comply with USP <1058>.
Resources
Data integrity requirements are tightening.
How do you keep up?
When surveyed, 84% of laboratories classify data integrity as “very” or “extremely” important. This attitude is not surprising, because regulators are now looking at all laboratory operations—including analytical instrument qualification—from a data integrity perspective.
In this era of evolving data integrity focus, FDA 483 reports demonstrate that laboratories are being cited because their controls do not prevent data manipulation.
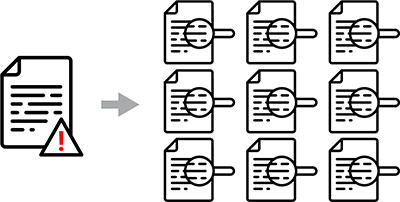
ALCOA versus ALCOA+
To satisfy data integrity requirements, your lab must first be compliant with core ALCOA principles:
- Attributable. Work is attributable to the person who performed it, typically through audit trails and a unique user login.
- Legible. Data are recorded in a permanent, readable format (ideally paperless).
- Contemporaneous. The data are recorded at the time the work is performed, which can be verified through electronic timestamps
- Original. All data—including metadata—are original or a certified true copy.
- Accurate. Errors, corrections, deviations, amendments, and repeat work are recorded.
However, there is an increased regulatory expectation for labs to comply with the additional requirements associated with ACLOA+ definitions, particularly analytical instrument qualification:
- Complete. All qualification tests and data, including repeat tests, are recorded and available for review.
- Consistent. Date/timestamps are applied in protocols and associated work.
- Enduring. Qualification protocols and reports are recorded in a secure, controlled, and enduring manner.
- Available. Qualification records are available throughout the required retention time.
Ensuring audit readiness by adapting to evolving challenges
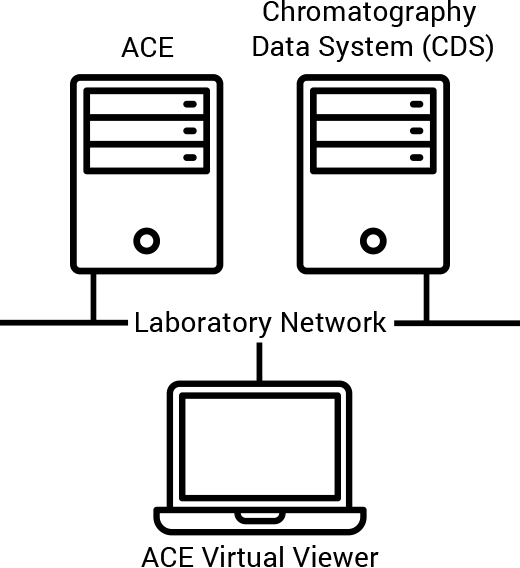
In response to the continued focus on data integrity, Agilent has upgraded our Automated Compliance Engine (ACE) software used for instrument qualification and reporting. Specifically, we enabled the installation of Network Distributed ACE within laboratory networks. This change ensures that all data stays within the lab network under IT policies and control.
Our new capability supports more advanced features. These include electronic qualification reports and plans, automated data traceability, electronic data transfer, and onsite harmonization of instrument qualification. It also allows you to configure protocols to satisfy USP <1058> requirements, so you can focus on your science while knowing that your lab is audit ready.
Resources
White paper
Case study
Brochures
Technical overview
Video
Flyer
What does lab-wide analytical instrument qualification (AIQ) look like?
Most labs contain technologies and instruments from different vendors. But when individual vendors perform qualification of ”their“ instruments, it creates a fragmented approach to lab-wide qualification that must be defended during audits and inspections. USP <1058> provides the framework for documenting that instruments are suitable for their intended use. However, it does not explicitly state what needs to be done.
As a result, regulated laboratories and instrument vendors tend to interpret USP <1058> requirements differently. Defending the scientific validity and data integrity of different approaches during audits only adds to the risk.
Some use paper-based protocols, while others manually transcribe data into an Excel or PDF template; still others use fixed protocols that cannot test range of use.
To comply with USP <1058> requirements, your AIQ procedure should support vendor and OEM instrument qualification aligned with your IT policy. It should also:
- Apply electronic audit trails and approvals.
- Be configurable for different user requirements.
- Enable electronic data transfer and secure data traceability.
- Facilitate harmonization of approach.
- Include calibration certificates and training records.
- Move away from higher-risk manual or Excel-based protocols.
- Support data retention.
- Test range of use.
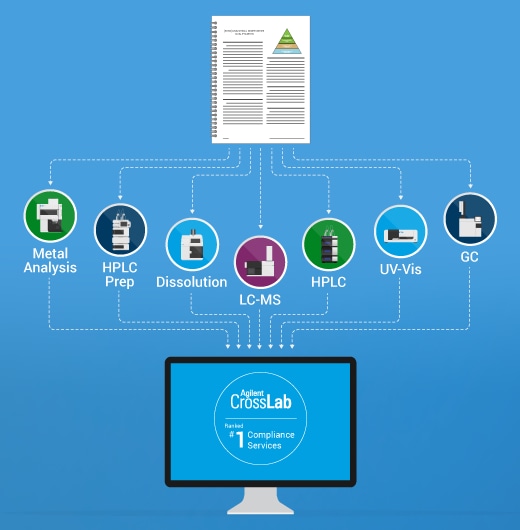
Moving to an approach that is independent of each vendor’s software can enable harmonization and remove the audit risk of fragmented qualification. Agilent qualification services align with all of these criteria, and support Agilent and non-Agilent instruments across chromatography, spectroscopy, elemental, and dissolution platforms. That means your lab can harmonize instrument qualification, simplify audit preparation, and focus on what matters.
Studies in the pharmaceutical industry show that for process validation, the largest single cost saving (approximately 30%) was achieved through the standardization and harmonization of approach. The harmonization of analytical instrument qualification within the laboratory, where the same principles apply, should produce similar benefits.
Resources
Brochures
Case study
Technical overview
Why is computer system validation so important?
As part of the evolving data integrity focus, regulators are closely examining software systems. In particular, auditors are looking for instances where software configuration and use may create data integrity issues. Confirming that a lab’s software configurations do not create data integrity risks is an essential part of computer system validation

How this new focus impacts your lab
Past audits may have looked for evidence of data integrity breaches. The current trend is for auditors to look at the “opportunity component” of Cressey’s fraud triangle—specifically, does the configuration prevent breaches? If your lab receives an FDA Warning Letter citing one of your computer systems, the remediation requirements will likely expand to include all of your systems.
For these reasons, computer systems must be validated for intended use, which is how the software is configured to meet regulatory requirements. That way, any deficiencies and risks will be documented (and corrected), and your lab can avoid the costs associated with regulatory action.
Software validation requirements are based on two primary approaches:
- GAMP 5—the risk-based approach and associated V model
- Electronic Records and Electronic Signatures (ERES)—21 CFR Part 11 for the U.S. and equivalent requirements for other regulatory agencies (such as Annex 11 in Europe)
What about computer system validation (CSV)?
Computer system validation (CSV) ensures that the intended workflows, including software use, are rigorously tested to identify and correct any issues. Although required for pharmaceutical and other regulated industries, it is often perceived as a cost that can delay implementation of essential new software.
By partnering with Agilent and applying Agilent CSV starter kits—which are based on the GAMP 5 model—your lab can save both time and money. In fact, an Agilent CSV starter kit can reduce implementation time by 50% or more, leaving you with more time for scientific analysis.
Resources
Flyers
Recorded seminar
Brochure
Self-qualification
To reduce downtime and “apparent” costs, many labs perform their own operational qualification (OQ) of analytical instruments. However, in-house protocols and calculations are typically paper- or transcription-based, which is time consuming, can be subjective, and introduces added risks. What’s more, procedural control may be the only data integrity safeguard available.
Does your lab perform in-house qualification?
The Agilent Partner ACE program can train and license your staff to use Agilent Automated Compliance Engine (ACE) software. Agilent-trained engineers use this software to deliver our paperless analytical instrument qualification (AIQ) services, which have gained top honors in independent surveys.
By partnering with Agilent, your qualification team can:
- Reduce the data integrity risks of paper- and Excel-based protocols.
- Automate and simplify your qualification workflow.
- Ensure that your qualification satisfies user requirements.
- Avoid the costs of regulatory action or developing electronic protocols in-house.
- Satisfy USP <1058> requirements.
- Harmonize qualification across analytical technologies.
After your team gains confidence, they can continue their education by purchasing flexible ACE credits from Agilent University.
Self-validation
Many regulatory data integrity findings focus on deficiencies in software configuration and use that could have been identified—and corrected—if the software had been validated. However, the skills required to efficiently validate software programs are complex. These skills include data integrity knowledge, software expertise, and regulatory understanding—including electronic records and signatures.
Concern about acquiring these skills may cause your lab to fall back on “legacy” software systems, while adding procedural layers to enhance data integrity. Even so, this is only a temporary fix.
Agilent GAMP 5-based CSV consultancy services make CSV easier through our CSV starter kits, which can reduce the validation life cycle by up to 50%. That’s time your lab could be spending on its science.
Our CSV specialists support:
- Audits and inspections of your current system (to identify risks).
- Compliance training and education for your staff.
- Starter kit documentation to your user requirements and needs.
- Custom procedure writing to support system use and SOPs.