Access Agilent eNewsletter July 2016

New LC/TQ database and method for central carbon metabolites
Christine Miller, Agilent Omics Market Manager
Discovery metabolomics is a powerful tool for identification of previously uncharacterized reactions, but many of the most striking “metabolic phenotypes” are changes in known pathways and intermediates. Numerous research questions depend upon the quantitation of known metabolites. The relative quantitation of central carbon metabolites is no exception. Agilent collaborators at the University of Toronto recently developed a liquid chromatography/triple-quadrupole (LC/TQ) mass spectrometry database and method to accelerate the process.
Although you can perform both discovery and follow-up analyses with accurate-mass time-of-flight (Q-TOF) LC/MS(TOF) and quadrupole-TOF instruments, the sensitivity, robustness, and straightforward methodology of LC/TQ mass spectrometry is ideally suited for targeted metabolomic analyses.
Reproducible analyses of large numbers of metabolites in a single injection
The Rosebrock and Caudy laboratories at the University of Toronto actively develop new experimental and analytical methods to enable high-throughput and high-content biology, with a focus on mass spectrometry based metabolomics. They recently developed a method for the relative quantitation of central carbon metabolites using dynamic multiple reaction monitoring (dMRM) on an Agilent 6460 Triple Quadrupole LC/MS system. dMRM uses retention-time scheduling of MRM transitions to allow you to analyze more metabolites in a single run without sacrifice of data quality.
“Dynamic MRM analysis lets us perform targeted analysis of many analytes in a single run, giving us a powerful window into metabolic phenotype,” according to Amy Caudy.
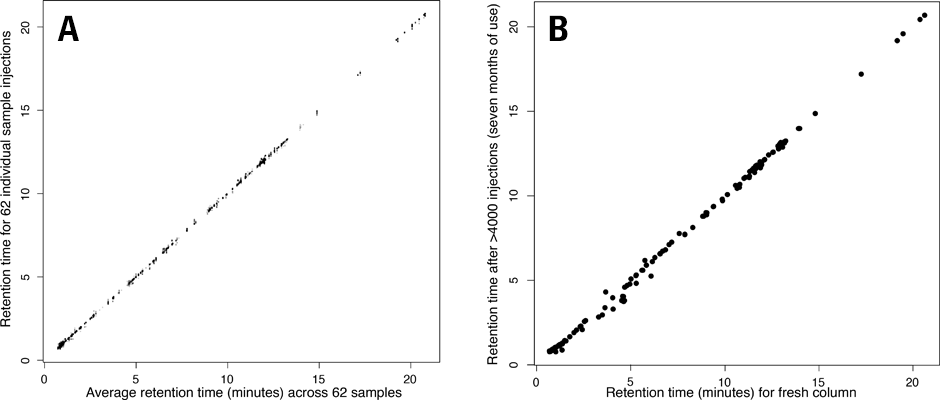
Figure 1. Chromatographic retention times are stable over thousands of injections. Shown are retention times for a set of known compounds.
Stable retention times enable large-scale studies
Rosebrock and Caudy developed a highly reproducible and robust ion-pair reversed-phase chromatographic method to separate the acidic central carbon metabolites, which included amino acids, citric acid cycle intermediates and other carboxylic acids, nucleobases, nucleosides, phosphosugars, and fatty acids. This ion-pairing method is reproducible between instruments and stable across time. Only very small retention time (RT) shifts have been observed in the analysis of large sample sets with thousands of injections (Figure 1).
In Figure 1A note the retention time deviation versus average retention time for 62 different biological samples (extracts of yeast cells) analyzed over the course of three days. Retention times for analytes in a single aliquoted sample, analyzed before and after an intervening 4000 injections of biological samples across many months is displayed in Figure 1B.
“The ability to generate data with minimal retention time shift across thousands of injections has enabled us to ask biological questions comprising large numbers of samples that were previously unapproachable,” says Adam Rosebrock.
Learn more about the Rosebrock and Caudy lab results in Application Note 5991-6449EN and this Agilent webinar.
dMRM database saves significant method development time
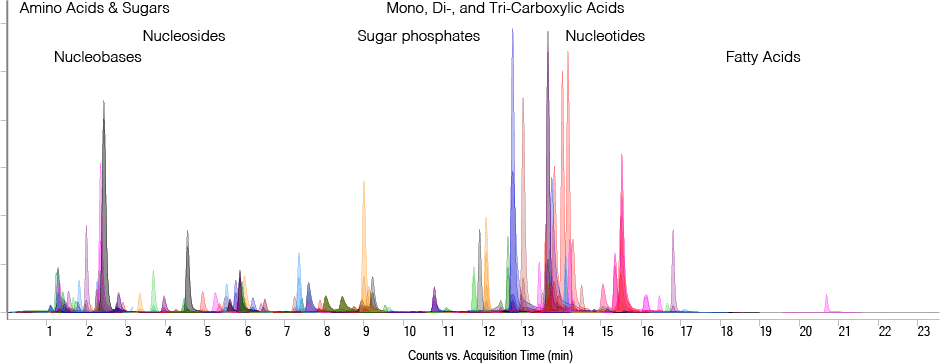
Figure 2. Overlaid MRM chromatograms of more than 100 metabolite standards at 5 ng on-column. LC elution ranges of metabolite classes are indicated.
Depending on the size of the target metabolite list, method development can be very time-consuming and requires the availability of a wide range of standards to optimize MRM transitions and retention times. The dynamic MRM database developed by the Rosebrock lab includes more than 215 metabolites from central carbon metabolism and provides Chemical Abstracts Service (CAS) identifiers, MRM transitions, retention times, and compound-specific MS parameters. In conjunction with the ion-paired reversed-phase chromatography method, the database enables sensitive detection of analytes across a wide dynamic range (Figure 2).
Read more about the database and method in Technical Note 5991-6482EN.
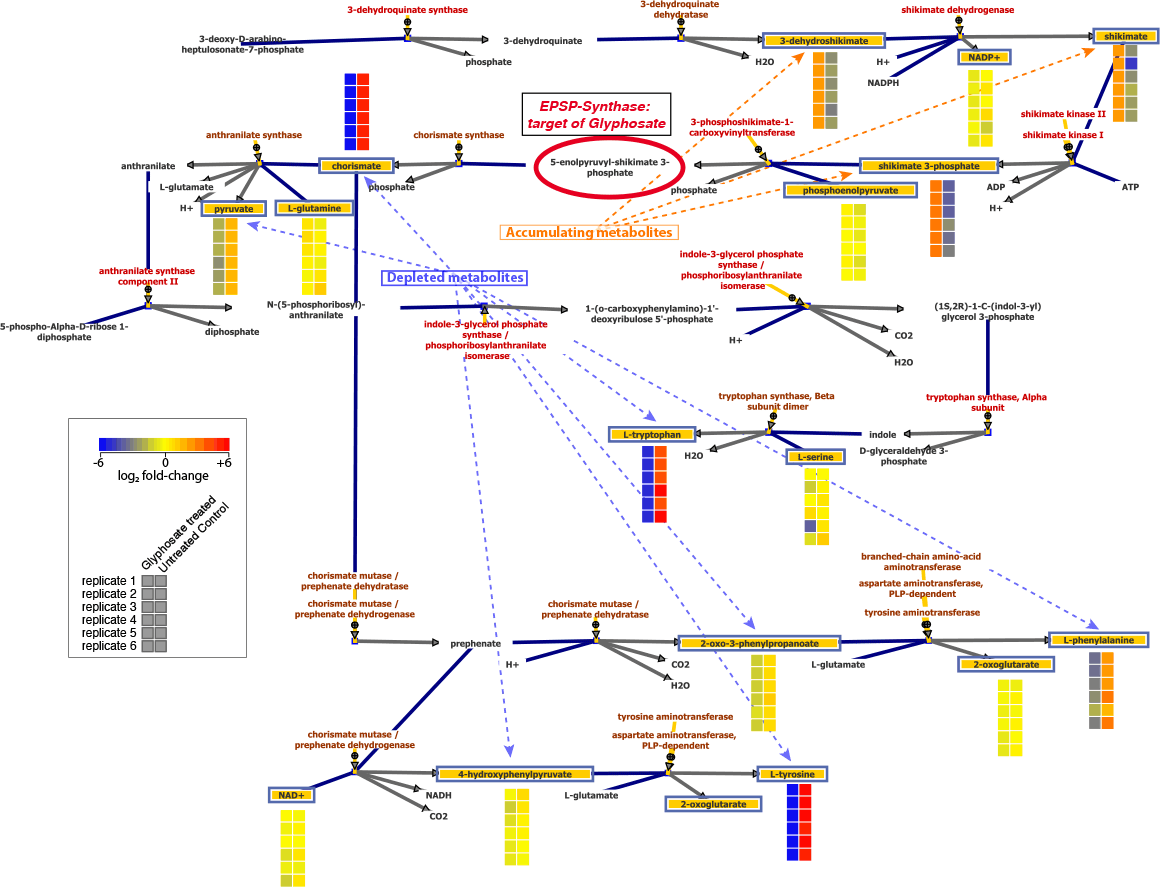
Figure 3. In E. coli treated with glyphosate, the pattern of increased and decreased metabolite levels upstream and downstream of EPSP synthase demonstrates the site of enzyme inhibition.
Seamless differential and pathway analysis
The targeted nature of this TQ-based method produces data that are implicitly associated with compound names and database identifiers, which enables seamless downstream differential and pathway analyses. This analytical method was used to assess relative changes in E. coli central carbon pathway metabolites after treatment with glyphosate (Figure 3). The results showed large increases in shikimate (shikimic acid) and 3-phosphoshikimate, while chorismate and all three aromatic amino acids were significantly decreased in concentration. Broad metabolite coverage of this library enables pathway-level analysis and characterization of a wide range of biochemical dysregulation.
The Metabolomics dMRM Database and Method based on this work are now available from Agilent. This solution allows any research lab to rapidly implement targeted analyses for central carbon metabolism, to minimize method development time and maximize research time.
Find out more about the Agilent Metabolomics dMRM Database and Method. Then contact your Agilent representative to put this application to work in your lab.
For Research Use Only. Not for use in diagnostic procedures.
Stay informed about the applications that are important to you
Subscribe to Access Agilent
Our free customized
monthly eNewsletter
All articles in this issue
 Make your Q-TOF LC/MS analyses faster, easier, and more productive—regardless of your application
Make your Q-TOF LC/MS analyses faster, easier, and more productive—regardless of your application Agilent J&W DB-624UI capillary GC column excels for challenging applications with active compounds
Agilent J&W DB-624UI capillary GC column excels for challenging applications with active compounds Tip: Using enzymes in dissolution testing of gelatin capsules
Tip: Using enzymes in dissolution testing of gelatin capsules InfinityLab Poroshell 120 columns maximize LC workflow efficiency
InfinityLab Poroshell 120 columns maximize LC workflow efficiency Emerging life science applications of FTIR Imaging: Providing spatially resolved molecular information in disease research
Emerging life science applications of FTIR Imaging: Providing spatially resolved molecular information in disease research New LC/TQ database and method for central carbon metabolites
New LC/TQ database and method for central carbon metabolites Agilent TQ systems enable ground-breaking research on use of metabolite biomarkers to predict a common pregnancy complication
Agilent TQ systems enable ground-breaking research on use of metabolite biomarkers to predict a common pregnancy complication Analysis of PEGylated proteins with Agilent AdvanceBio SEC columns
Analysis of PEGylated proteins with Agilent AdvanceBio SEC columns Structural elucidation of in-process impurities using high resolution LC/MS/MS
Structural elucidation of in-process impurities using high resolution LC/MS/MS Removal of lipids for the analysis of toxicological compounds in plasma by LC/MS/MS
Removal of lipids for the analysis of toxicological compounds in plasma by LC/MS/MS Quick, accurate, cost-effective measurements of veterinary drugs in meat with Agilent MS/MS, UHPLC, and Q-TOF
Quick, accurate, cost-effective measurements of veterinary drugs in meat with Agilent MS/MS, UHPLC, and Q-TOF
Figure 1
Chromatographic retention times are stable over thousands of injections. Shown are retention times for a set of known compounds.
Figure 2
Overlaid MRM chromatograms of more than 100 metabolite standards at 5 ng on-column. LC elution ranges of metabolite classes are indicated.
Figure 3
In E. coli treated with glyphosate, the pattern of increased and decreased metabolite levels upstream and downstream of EPSP synthase demonstrates the site of enzyme inhibition.