
It is only natural to be nervous about preparing for a regulatory inspection—even when you have a strong data integrity (DI) program in place. Preparation is easier when you understand why DI inspections are becoming more commonplace today, what the auditors are looking for, and how to answer their questions.
This article summarizes the typical scope and focus of a DI inspection and provides six best practice tips for simplifying, expediting, and surviving your next DIO (data integrity observation) audit.
DI issues are drawing more attention
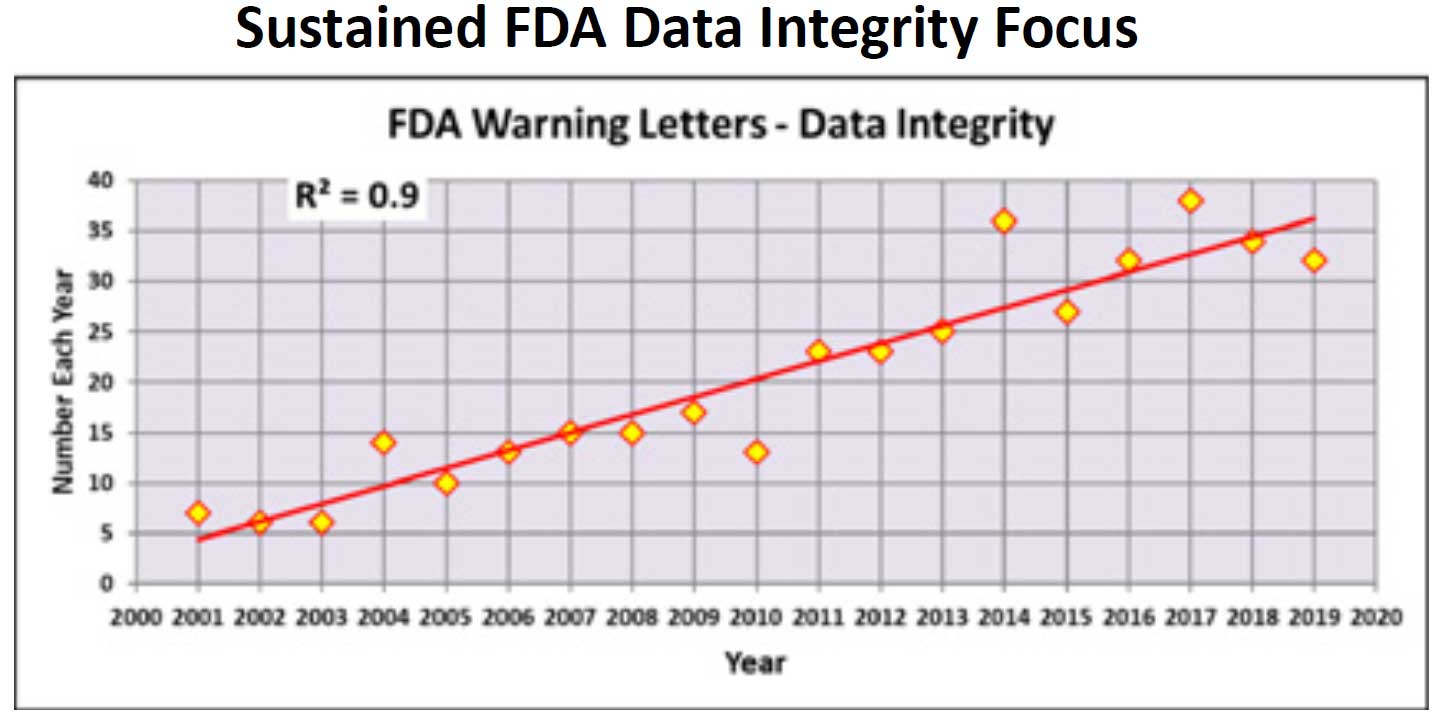
Figure 1.FDA Data Integrity Focus
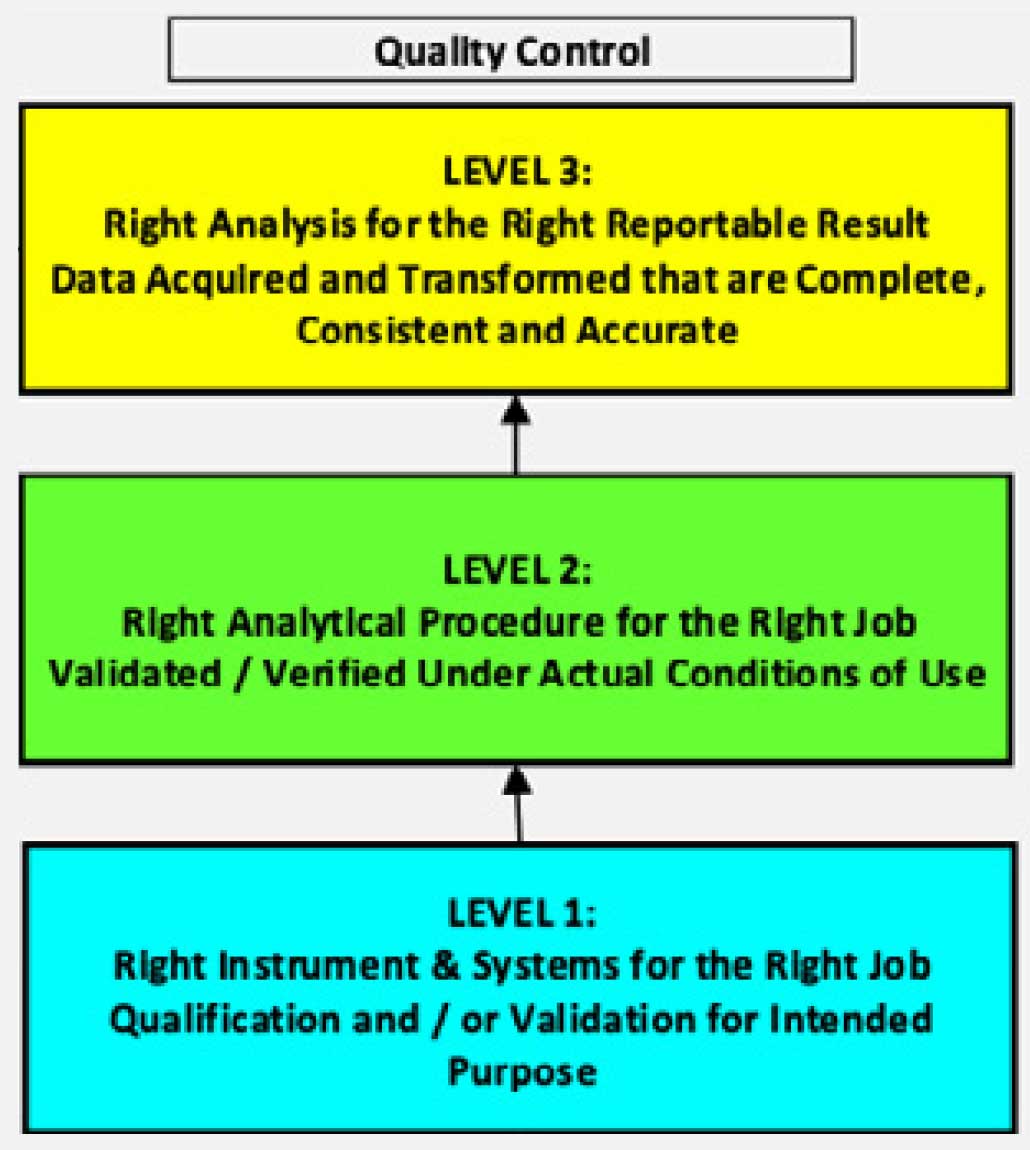
Figure 2.Pharmaceutical quality system: Quality Control
Data integrity has come under increasing scrutiny by regulatory agencies over the past 20 years, and inspectors are focusing much of their attention on CDS issues such as unofficial testing, deletion of data, failure to protect electronic records, integration into compliance, and the selective invalidation of out-of-specification (OOS) results.
No two inspections are the same, but there is often a common scope. The focus areas of a typical FDA inspection are illustrated below. The first level has to do with validation of the instruments and information management system used at the laboratory. All equipment must be kept in calibration against reference standards. The second level includes the procedures and methods they are used for. The right analytical procedure must be applied to the analysis, and the methods must be validated and verified under their actual conditions of use. At the top level is the analysis of the sample. The correct analysis must be applied to obtain the relevant result.
A failure at any level invalidates the level(s) above it. The right analysis cannot be done if the right analytical procedure has not been established, and neither the analysis nor the procedure can be performed if the instrumentation is not validated and in calibration.
1. Enable audit trails on your CDS.
Every available audit trail should always be enabled on your CDS software. Electronic audit trails keep a contemporaneous record that is easy to review and help support thorough investigations. In addition, put procedures in place for regular audit trail reviews as a part of any approval of results to verify that the reported result is valid.
2. Always use e-signatures.
Use of electronic signatures in your CDS software will speed up inspections and approvals. Each user must have their own unique account that is never shared, and lock data after review, preventing additional changes that could undermine the trustworthiness of the reported results. Use multiple signature levels, control the access to the application of signatures, and enforce the order of signatures to support the documented workflow.
3. Grant access cautiously.
Compliance with 21 Code of Federal Regulations (CFR) Part 11 FDA regulations requires that user access to systems must be controlled. When setting up the system, it is important to grant permissions to users based on their job role and the specific workflow. Users should be given access only to those features required for them to complete their job. The best practice is to establish group permissions, based on specific roles, that the system administrator can easily maintain and review.
4. Ensure that all roles are well defined.
To prevent conflicts of interest, clearly define and document the access privileges that your data system users have, including segregation of roles to prevent conflict of interest. Start by defining each group of users and actions each group of users should be able to perform based on their role. Use these definitions to configure system permissions and be ready to show that they are in sync with the documentation.
5. Expect to be asked about aborted runs.
The inspector will be looking to see how the system is used in practice. Short or aborted runs in the system or orphan samples/orphan data may indicate a problem with some analysis, and inspectors want to see how you handle those problems. For example, consider what happens when an incorrect number is typed into a sample weight. Upon seeing this, an inspector will demand thorough documentation of how the issue was handled.
6. Make second-person reviews routine.
The scope of the second person review should cover everything from the sampling to reporting of results. It is important that this is performed correctly and diligently by an experienced analyst. The reviewer should focus on sample preparation records and on manual data entered into the CDS sequence file (e.g., weights, purities, or dilutions, and should ensure that peak integration is correct, especially manual integration.
One last piece of general advice to ensure a smooth inspection process: only answer the question asked, but if you are asked something that has verifying documentation to support the answer, have that documentation ready. This will make the process go much more smoothly and prevent antagonizing the inspector or raising suspicions that you have something to hide.
OpenLab CDS: Features that facilitate regulatory compliance
OpenLab CDS is a chromatography data system that streamlines compliance with data integrity regulatory requirements in multiple ways. For example, it offers:
- Secure central storage
- Avoidance of transcription errors through the Custom Calculator tool, which ensures that all the calculations take place within the chromatography system not manually or automatically exported to external spreadsheet applications
- Audit trail functionality covering the whole system, with technical controls within the audit trail to highlight data changes and deletions to facilitate the review process
- Functionality within the CDS application to document that audit trail entries have been reviewed
Learn more
For more detailed advice about DI inspections, check out the related content below.
